
In this project, I developed a new method for detecting hydrogen-bond (H-bond) interactions in ProLIF (Protein-Ligand Interaction Fingerprints). ProLIF, based on RDKit and MDAnalysis, is a python package to calculate protein-biomolecular interactions for an molecular dynamic trajectory. Before this project, calculating H-bond interactions with ProLIF required a complete topology, including both heavy atoms and explicit hydrogens (explicit H-bond method). Here, using only heavy atoms (and implicit hydrogens), we are able to recall the same H-bond interactions detected by the explicit H-bond method while also identifying ones that were missed. This method can help users more easily calculate the H-bond interactions for experimental and computational structures without additional protonation steps. Moreover, I provided a function to parse PDB files with non-standard residues, and this can also be used to modify the residues into user-defined protonation states. Finally, our implicit H-bond method was compared and validated with the explicit H-bond method using PINDER (2024-02) and PLINDER (2024-06/v2) datasets.
![]()
![]()
Contributor: Yu-Yuan (Stuart) Yang (@yuyuan871111)
Mentors: Cédric Bouysset (@cbouy, @cbouysset), Valerij Talagayev (@talagayev)
Organization: MDAnalysis
This is an archive of my GSoC 2025 final report [original link].
What I did during this summer?
During my GSoC project, I focused on three key deliverables: Protein helper function, Main function of calculating implicit H-bond interactions, and Method validation. Here, I demonstrate how to install ProLIF in your environment and use these new functions (ProteinHelper and ImplicitHBDonor/ImplicitHBAcceptor) in ProLIF.
Installation and quick start
Before running the below examples, please follow the below guide to install the specific version of ProLIF (GSoC implicit H-bond branch). For the general usage, please follow this guide to install ProLIF and check this tutorial.
# create a virtual environment with conda
conda create --name implicit_hbond python=3.11 -y
conda activate implicit_hbond
# install prolif and requirements in your conda environment
pip install git+https://github.com/chemosim-lab/ProLIF.git@gsoc_implicit_hbond
pip install rdkit # required for ProLIF
pip install gemmi # required for the below example
pip install notebook # if you want to run the script in jupyter notebook (interactively)
pip install py3Dmol # 3D visualization within jupyter notebook
Then, you will need to create a jupyter notebook and start it to run the script below.
jupyter notebook
Protein Helper (parse PDB files with non-standard residues)
Please download the 5da9.pdb (mirror archive) from RCSB PDB to your local folder. It is an example of protein-ligand interactions. In this example, MSE is a non-standard residue. MG and AGS are ligands for the complex, whose bond orders can be corrected by using the same ProteinHelper with SMILES strings.
You might need to modify the path for the files in the below script. [Notebook version] [Download here]
# import the required packages
import MDAnalysis as mda
import prolif as plf
from prolif.molecule import Molecule
from prolif.io.protein_helper import ProteinHelper
# initialize the protein helper function
protein_helper = ProteinHelper(
[
{
"MSE": {"SMILES": "C[Se]CC[CH](N)C=O"},
"MG": {"SMILES": "[Mg++]"},
"AGS": {"SMILES":
("c1nc(c2c(n1)n(cn2)[C@H]3[C@@H]"
"([C@@H]([C@H](O3)COP(=O)(O)OP(=O)(O)OP(=S)(O)O)O)O)N")}
}
]
)
# the current version of ProLIF cannot split the molecule directly
# thus, MDAnalysis are used for splitting protein and ligand into separate files
u = mda.Universe("./test_data/5da9.pdb")
u.select_atoms("protein or water").write("./test_data/5da9_protein.pdb")
u.select_atoms("not protein and not water").write("./test_data/5da9_ligand.pdb")
# read the PDB using protein helper
protein_mol = protein_helper.standardize_protein("./test_data/5da9_protein.pdb")
ligands = protein_helper.standardize_protein("./test_data/5da9_ligand.pdb")
# check the residues with plf.display_residues
# this will help you check the bond order of the residues used for calculating fingerprint
plf.display_residues(protein_mol, slice(0, 20), sanitize=False)
plf.display_residues(ligands, slice(0, 20), sanitize=False)
# here, we focused on the AGS ligand at chain A.
ligand = Molecule(ligands[1])
# let’s calculate implicit H-bond interactions
fp = plf.Fingerprint(["ImplicitHBDonor", "ImplicitHBAcceptor"], count=True)
fp.run_from_iterable([ligand], protein_mol, progress=False)
fp.to_dataframe() # print result
# 2D visualization
view = fp.plot_lignetwork(ligand, kind="frame", frame=0, display_all=False)
view
# 3D visualization
view = fp.plot_3d(ligand, protein_mol, frame=0, display_all=True)
view
Here is the expected result for 3D visualization: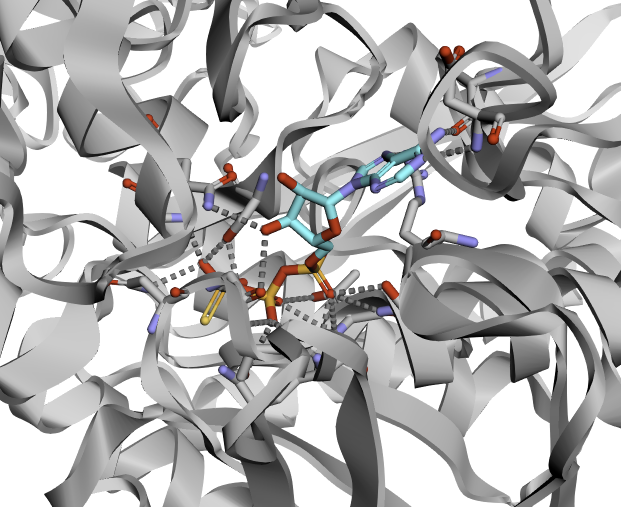
Implicit H-bond interactions (comparison between experimental and computational structures)
The example shows how to calculate implicit H-bond interactions for a protein-RNA complex from RCSB PDB (experimental: 8AW3, mirror archive) and AlphaFold3 webserver (computational: 8AW3, mirror archive). The cif templates for RNA residues (A, U, C, G; mirror archive) can be found on RCSB PDB. It is noted that the computational structure is in cif format, which is not supported by MDAnalysis 2.9.0 (could be supported in the future version). Thus, we currently read it via gemmi and converted it into pdb format.
You might need to modify the path for the files in the below script. [Notebook version] [Download here]
# import required packages
import prolif as plf
import MDAnalysis as mda
import gemmi
from MDAnalysis.analysis import align
from prolif.io.cif import cif_template_reader
from prolif.io.protein_helper import ProteinHelper
# get the protein helper
protein_helper = ProteinHelper(
[
{"ZN": {"SMILES": "[Zn++]"}},
cif_template_reader("./test_data/templates/A.cif"),
cif_template_reader("./test_data/templates/U.cif"),
cif_template_reader("./test_data/templates/C.cif"),
cif_template_reader("./test_data/templates/G.cif"),
]
)
# split the experimental structure into protein and ligand
u = mda.Universe("./test_data/8aw3.pdb")
u.select_atoms("protein or water").write("./test_data/8aw3_protein.pdb")
u.select_atoms("not protein and not water").write("./test_data/8aw3_ligand.pdb")
# read the protein and ligand (and correct the bond orders)
protein_mol = protein_helper.standardize_protein("./test_data/8aw3_protein.pdb")
ligand = protein_helper.standardize_protein("./test_data/8aw3_ligand.pdb")
# run to get fingerprint for the experimental structure
fp = plf.Fingerprint(["ImplicitHBDonor", "ImplicitHBAcceptor"], count=True)
fp.run_from_iterable([ligand], protein_mol, progress=False)
fp.to_dataframe() # print result
# read the computational structure with gemmi and convert it into pdb
structure = gemmi.read_structure("./test_data/examplefold_pdb_8aw3_model_0.cif")
structure.write_pdb("./test_data/examplefold_pdb_8aw3_model_0.pdb")
# read the computational structure with mdanalysis and separate the protein and ligand
u_comp = mda.Universe("./test_data/examplefold_pdb_8aw3_model_0.pdb")
# align the structure to the reference using MDAnalysis for better visualization
# (not affect the calculation of fingerprint)
align.alignto(u_comp, u, select="backbone and name CA and resid 183-221")
u_comp.select_atoms("protein or water").write(
"./test_data/examplefold_pdb_8aw3_model_0_protein.pdb"
)
u_comp.select_atoms("not protein and not water").write(
"./test_data/examplefold_pdb_8aw3_model_0_ligand.pdb"
)
# read with protein helper
protein_mol_comp = protein_helper.standardize_protein(
"./test_data/examplefold_pdb_8aw3_model_0_protein.pdb"
)
ligand_comp = protein_helper.standardize_protein(
"./test_data/examplefold_pdb_8aw3_model_0_ligand.pdb"
)
# run to get fingerprint for the computational structure
fp_comp = plf.Fingerprint(["ImplicitHBDonor", "ImplicitHBAcceptor"], count=True)
fp_comp.run_from_iterable([ligand_comp], protein_mol_comp, progress=False)
fp_comp.to_dataframe() # print results
# Visualization in parallel (3D)
# left: experimental; right: computational
comp3D = fp.plot_3d(ligand, protein_mol, frame=0)
other_comp3D = fp_comp.plot_3d(ligand_comp, protein_mol_comp, frame=0)
view = comp3D.compare(other_comp3D, display_all=True, remove_hydrogens=False)
view
Here is the expected example for 3D visualization: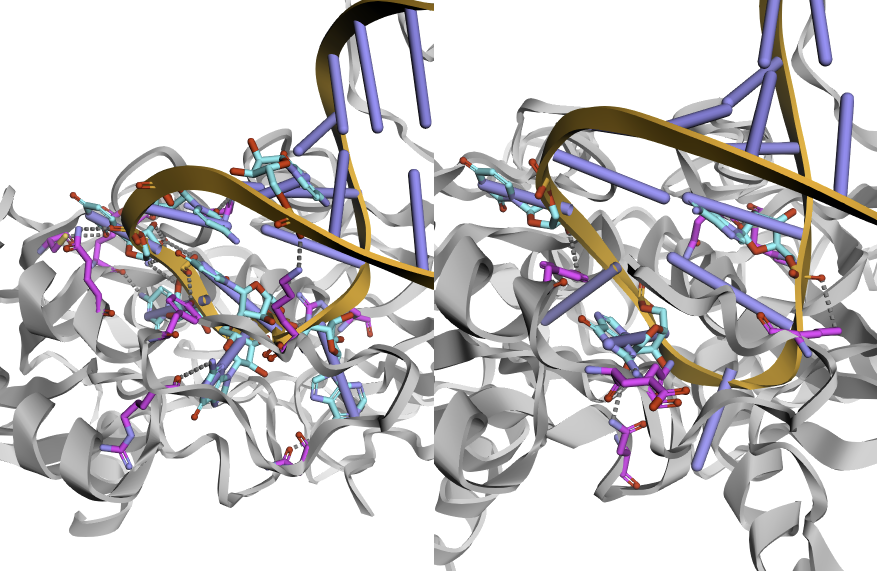
Benchmark and validation (comparison with the explicit H-bond method)
Since the implicit H-bond method was a new function of ProLIF, I compared it to the existing explicit H-bond method using PLINDER (protein-ligand complexes) and PINDER (protein-protein complexes) datasets. Also, I used those datasets to fine-tune some geometric parameters used in the implicit H-bond method (more details in the week 10 blog post). Here, to be simple, I demonstrate a case (PLINDER: 5da9) to compare the results from implicit and explicit H-bond methods. For further details of our validation using a batch of data (PLINDER/PINDER validation and testing datasets), please check the repository and data archive below. The protonated pdb files in the validation and testing datasets of PLINDER and PINDER are provided below in Zenodo archive. The results of the implicit and explicit H-bond calculations are also saved in Zenodo archive; however, some data are not available now due to file reading errors (mostly due to poor definitions or predictions of connectivity and bond orders in the protonated molecule).
- Repository: https://github.com/yuyuan871111/GSoC2025_Hbond_PM/tree/main/validation
- Data archive on Zenodo: https://doi.org/10.5281/zenodo.16950708
- Mirror archive of Zenodo on Google Drive: 03_Validation_Release
You might need to modify the path for the files in the below script. [Notebook version] [Download here]
import os
import prolif as plf
from prolif.io.protein_helper import ProteinHelper
from prolif.plotting.complex3d import Complex3D
from prolif.molecule import Molecule
from prolif import sdf_supplier
from rdkit import Chem
protein_helper = ProteinHelper(
templates=[
{
"MSE": {"SMILES": "C[Se]CC[CH](N)C=O"},
}
]
)
# test case
test_case_dir = "./test_data/5da9__1__1.A_1.B__1.E_1.F"
# read the protonated protein structure
mol = Chem.MolFromPDBFile(
f"{test_case_dir}/receptor_protonated.pdb",
sanitize=False,
removeHs=False,
proximityBonding=True,
)
protein_mol = Molecule.from_rdkit(mol)
fixed_protein_mol = protein_helper.standardize_protein(protein_mol)
# read the ligand (sdf format)
ligands = []
for ligand_sdf in os.listdir(test_case_dir): # noqa: PTH208
if ligand_sdf.endswith("_protonated.sdf"):
ligands.extend(sdf_supplier(f"{test_case_dir}/{ligand_sdf}"))
ligand = ligands[0] # we focus on the non-metal ion ligand
# Explicit H-bond method
fp = plf.Fingerprint(["HBDonor", "HBAcceptor"], count=True)
fp.run_from_iterable([ligand], protein_mol, progress=False)
df = fp.to_dataframe().T
# 2D visualization
view = fp.plot_lignetwork(ligand, kind="frame", frame=0, display_all=False)
view
# read the same molecule but remove hydrogens
protein_mol_i = protein_helper.standardize_protein(
Molecule.from_rdkit(Chem.RemoveAllHs(protein_mol))
)
ligand_i = Molecule.from_rdkit(Chem.RemoveAllHs(ligand))
# Implicit H-bond method
fp_i = plf.Fingerprint(
["ImplicitHBDonor", "ImplicitHBAcceptor"],
count=True,
)
fp_i.run_from_iterable([ligand_i], protein_mol_i, progress=False)
df_i = fp_i.to_dataframe().T
# 2D visualization
view = fp_i.plot_lignetwork(ligand_i, kind="frame", display_all=False)
view
# Compare in parallel
comp3D = Complex3D.from_fingerprint(fp, ligand, protein_mol, frame=0)
other_comp3D = Complex3D.from_fingerprint(
fp_i, ligand_i, protein_mol_i, frame=0
)
# left: explicit; right: implicit
view = comp3D.compare(other_comp3D, display_all=True)
view
Here is the expected result (3D visualization):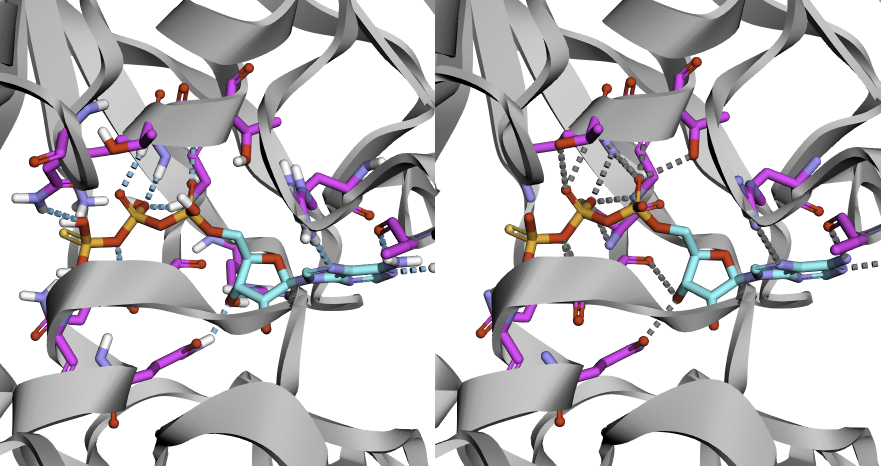
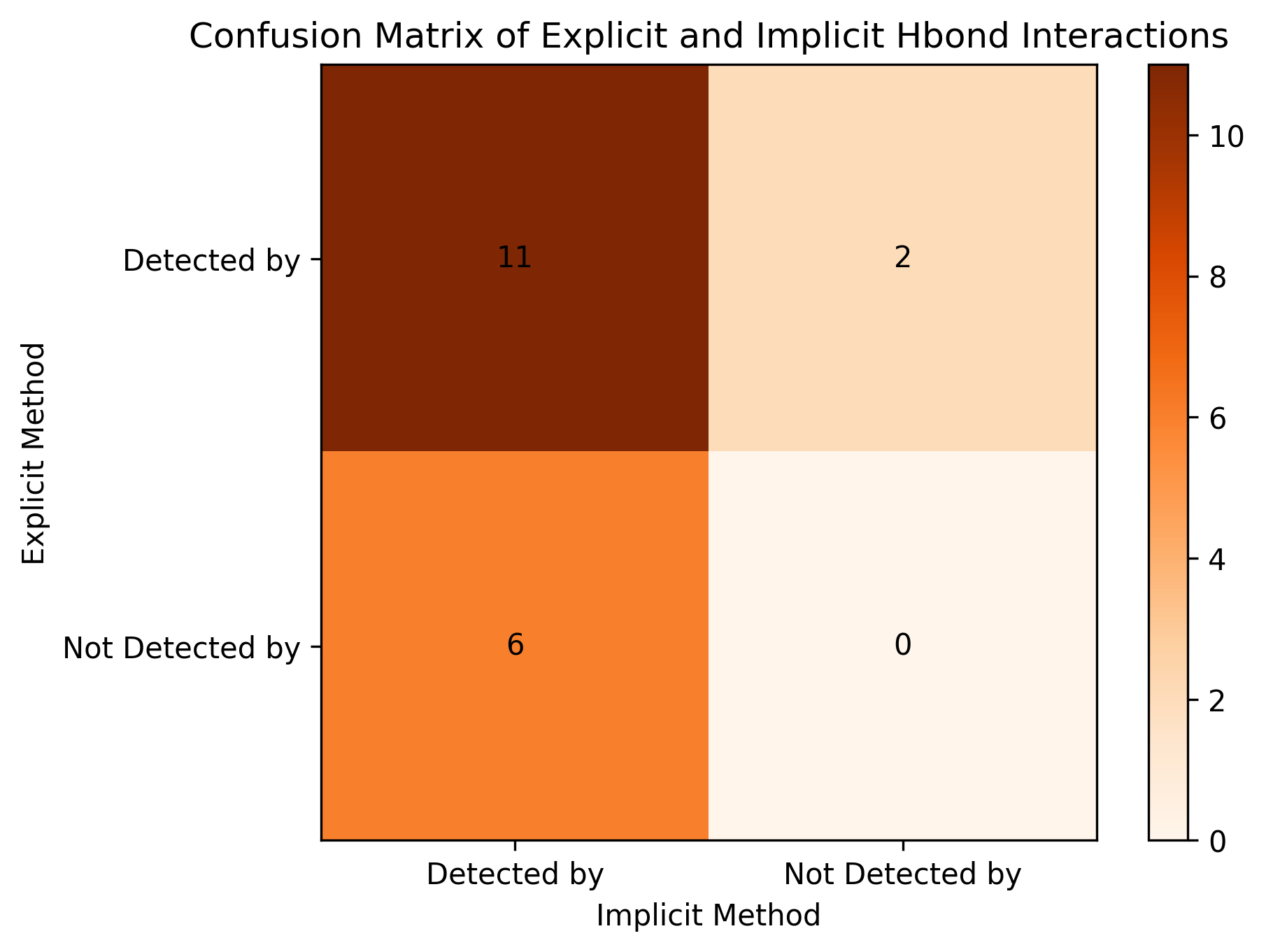
By comparing the calculated donor-acceptor pairs between explicit and implicit methods, we created a confusion matrix, shown in the below figure. We can also calculate the Tanimoto coefficient to check the similarity of the results between two methods. For this case, Tanimoto coefficient is 0.58, which is in an acceptable range. Most H-bond interactions are detected by both methods.
Pull requests (PRs)
Besides the deliverables, the PRs are the core of open-source software development, including three general steps: opening a PR, developing code, and being merged into the repository. During the stage of code development, there is a back-and-forth progress to improve the quality of code based on feedback from code reviewers. If you are interested in the details of my PRs, please see the PRs below.
Initially, I worked on ProLIF’s gsoc_implicit_hbond branch [link]. Once the development is complete, the branch will be merged into the main branch.
- Protein Helper function: PR#275 (merged into
gsoc_implicit_hbond) - Main function for the implicit H-bond method: PR#284 (merged into
gsoc_implicit_hbond) - Implicit H-bond method development into main ProLIF: PR#302 (going to be merged into
main)
Blogs
During my GSoC journey in the summer of 2025, I documented my progress and reflections through blogs. These posts not only shared my experiences but also served as user guides, emphasizing the significance of protein helper and implicit H-bond interactions. If you’re curious about what I accomplished and want a glimpse into what your own GSoC experience might look like, feel free to explore the links below.
- Introduction of my GSoC 2025 project, MDAnalysis, and ProLIF [GSoC 2025 - Week 0]
- Protein helper function fixed your bond orders of residues (molecules) [GSoC 2025 - Week 4]
- Have a better design of protein helper function during code reviews [GSoC 2025 - Week 6]
- What is the implicit hydrogen? Why need an additional interaction class? [GSoC 2025 - Week 8]
- Validation and Optimization: tuning parameters for the implicit H-bond interaction method using datasets [GSoC 2025 - Week 10]
- Development Wrap-Up: PINDER validation and comprehensive documentation [GSoC 2025 - Week 12]
Future work
First, in the current work, I used only validation and testing sets from PLINDER and PINDER for parameter tuning and testing. More data in PLINDER training (n ~= 420K) and PINDER training (n ~= 1.6M) sets can be used for further validation.
Second, two versions of the molecule are stored in ProLIF’s molecule object: “whole” molecule and molecule fragmented by residue. The former one is mainly used for visualization and the latter one is used for the fingerprint analyses. In this project, I developed the protein helper function to fix the bond order of the molecule fragmented by residues (that is, fix it at residue level) instead of the “whole” molecule. This can lead to a visualization error or discrepancies at the molecular level, but does not affect the calculation of the fingerprint (using residue-level information). Thus, further testing is required to enhance the user experience for visualizing the “whole” molecule in either a 2D or 3D plot.
Lessons learned during GSoC
Now, I can proudly say, “I am an open-source software contributor!”
At the start of my GSoC journey, I learned how to craft a strong proposal that clearly communicates ideas and solutions to specific challenges. I also experienced the thrill of getting my first PR merged into the codebase—after plenty of discussions and several rounds of code revisions, of course!
Throughout the development process, I came to appreciate the critical role of test functions (using pytest) in open-source software development. These tests are often overlooked but are essential for maintaining the integrity of the codebase. They ensure that contributions from multiple developers don’t inadvertently break or undo the work of previous contributors.
Additionally, I honed my skills in managing development environments more efficiently with tools like uv, poetry, ruff, and poe-the-poet. I also learned techniques to minimize code duplication, which is key to writing cleaner and more maintainable code.
Last but not least, documentation should always be simple and straightforward. For new users, their first impression often comes from the documentation, and it plays a crucial role in guiding them. A well-written, user-friendly document can encourage them to continue using the software and explore its features further. So, make the product shine and let the documentation speak for itself!
Acknowledgements
I want to thank my mentors, Cédric (@cbouy, @cbouysset) and Valerij (@talagayev). Their support and responsiveness were invaluable—they provided excellent guidance not only on coding but also on mindset and career development. I am lucky to have been mentored by them.
I would also like to thank MDAnalysis for welcoming me into the community and giving me the chance to contribute to such high-impact Python packages. A special thank you goes to the organization admins, Irfan (@IAlibay) and Jenna (@jennaswa), for their efforts in making this experience possible.
Finally, I extend my thanks to Google for hosting and sponsoring this program over the years, fostering the growth of open-source software and creating opportunities for contributors like me. I hope to become a mentor next year and support new contributors on their own journeys, just as my mentors supported me.
Note: The top picture was generated by Recraft V3 Raw with Handmade 3d style, aspect ratio: 16:9, generation count 4 (I picked one picutre out of four.), and prompts: “I am an open-source software contributor!!! I am excited to share my project on hydrogen bond interactions for structural biology and molecular dynamics.”. I then modified the image to replace the background with Google Summer of Code logos (compliance with Google Summer of Code Brand Guidelines).