![Have a better design of protein helper function during code reviews [GSoC 2025 - Week 6]](/images/blog_pics/gsoc-wk-6.png)
Well, two weeks ago, I developed my protein helper function. Before merging codes into the codespace, I will need to undergo a process “code review”.
During the code review period, the code style will need to be aligned to the main codespace, the file will be organized better, and the design of the functions will be improved.
After a few rounds of the code review, the code will be merged soon.
What specific things you will need to do during the code review?
Develop test functions
After developing the new functions, one thing need to do is to write the test functions. The test functions can check whether you ruin some original functions when building the new functions. Also, test functions ensure that your code remains robust and prevent issues caused by careless contribution from others.
Simple example
This is a simple test to check whether the constants are read correctly.
def test_standard_aa() -> None:
"""Test that STANDARD_AA contains the expected amino acids."""
from prolif.constants import STANDARD_AA
assert len(STANDARD_AA) == 26
assert len(STANDARD_AA["ALA"]) == 4
assert isinstance(STANDARD_AA["ALA"]["_chem_comp_atom"], pd.DataFrame)
Complicated example
pytest is a powerful package to create test functions. Here is a more complicated case for writing test functions. I used some decorator in pytest.
The below example is to use @pytest.fixture decorator. The fixture decorator can create a reliable context (input) for different test functions. Thus, you don’t need to repeatedly define your inputs in each test function.
@pytest.fixture(scope="module")
def XML_TEST_DATA() -> str:
"""Fixture to load the XML test data for altnames parsing."""
return (datapath / "standard_aa_name.xml").read_text()
def test_xml_parse_altnames(XML_TEST_DATA: str) -> None:
"""Test the XML parsing of alternative names for residues and atoms."""
resname_aliases, atomname_aliases = parse_altnames(XML_TEST_DATA)
assert isinstance(resname_aliases, dict)
assert isinstance(atomname_aliases, dict)
Another case is to use @pytest.mark.parameterize. If you want to tests different conditions of your function, it is helpful (to save your time to write the same codes).
from prolif.io.protein_helper import ProteinHelper
@pytest.mark.parametrize(
("resnames", "expected"),
[
({"HSD"}, "charmm"),
({"NASP"}, "amber"),
({"ASN1"}, "gromos"),
({"HISD"}, "oplsaa"),
({"CYS"}, "unknown"),
],
)
def test_forcefield_guesser(resnames, expected) -> None: # type: ignore
"""Test the forcefield guesser."""
# Test with a known forcefield
forcefield = ProteinHelper.forcefield_guesser(resnames)
assert forcefield == expected
Check code/linter style and add type hints
Good coding practices include maintaining a consistent code style and having type hints, even though these don’t affect the functionality or usability of the code. A consistent code style will increase the readability of code, which is especially important in a project with others.
Here is an example without a consistent code/linter style and type hints:
import pandas as pd
def getGenus(df):
Genus_name = list(df.index)
return Genus_name
from collections import deque
class TreeNode :
data = None
left = None
right = None
def __init__(self,data=None, left = None ,right = None) :
self.data = data
self.left = left
self.right = right
After improvement:
import pandas as pd
from collections import deque
def getGenus(df: pd.DataFrame) -> list:
"""Document for getGenus function"""
return list(df.index)
class TreeNode:
"""Tree Node document"""
def __init__(self, data: Any = None, left: Any = None, right: Any = None):
self.data = data
self.left = left
self.right = right
Can you tell the difference between two examples?
(Give you five seconds to think about it.)
.
..
…
….
…..
Here are what I found (you can check whether they are the same as your answers):
- In general, all imported packages should be on the top of the script (e.g.
import ...,from ...). - Spacing should be consistent. You can choose either 2 or 4 spaces but you need to make sure it is consistent in the whole file. Also, make sure there is a fixed number of space before and after operators like
=and seperators like,(e.g.,a, b, cinstead ofa , b,c). - Add document in your function. Use
""" ... """to write the document. When people use your functions, it is more easier to know how they work. - Add type hints in your function. You can accelerate your code development if you know the type of the data. Same as others. If the user know what data type of the output of a function, the user won’t spend lots of time to debug and check with the data. Thus, it would be better to have type hints.
- Remove redundant variable assignment. You might notice that
getGenusfunction is slightly different. The variable assignment “Genus_name = list(df.index)” is unnecessary. This can slightly speed up your code.
Actually, there is a list of rules to make your code consistent, such as PEP8, Flake8, isort, etc. Some tools, like Ruff, can manage those rules and automatically format the code with a consistent set of code/linter styles.
Improve the design of the function
Sometimes, you might have a major revision of your function. In my case, I will need to inverting the rolo of two functions (__init__ and standardize_protein).
Original protein helper function
In the original design of the protein helper function, the initialization requires a topology file of the molecule. The templates are provided during the standardization of the molecule. The output is stored in the protein helper function’s protein_mol.
from prolif.datafiles import datapath
from prolif.io.protein_helper import ProteinHelper
# read the protein pdb
protein_path = datapath / "implicitHbond/1s2g__1__1.A_2.C__1.D/receptor_hsd.pdb"
protein_helper = ProteinHelper(input_topology=protein_path)
# standardize the protein (using default template by assigning templates with None)
protein_helper.standardize_protein(templates = None)
# the output is stored at
protein_helper.protein_mol
The updated protein helper function
The new version of protein helper function is initialized using templates. The standardize_protein function is reusable for different proteins without requiring another protein helper function. See below example:
from prolif.datafiles import datapath
from prolif.io.protein_helper import ProteinHelper
# call a protein helper
protein_helper = ProteinHelper(templates = None)
# read and standardize the protein
protein_path = datapath / "implicitHbond/1s2g__1__1.A_2.C__1.D/receptor_hsd.pdb"
output_protein_mol = protein_helper.standardize_protein(input_topology=protein_path)
This design of protein helper function is more intuitive for users and is also more efficient. However, I didn’t come up with this design during development. My code reviewer (and my mentor), Cédric, proposed this idea, and I suddenly realized it was true. This experience highlights how the code review process can definitely improve the quality of the code.
Code review process
You can see my progress of code review here. The pull request was created at the start of June (week 2-ish), but the discussion, code modifications and new code reviews have been ongoing until now. The code has been iterated through several rounds.
First version of the protein helper function
New version of the protein helper function with further discussions

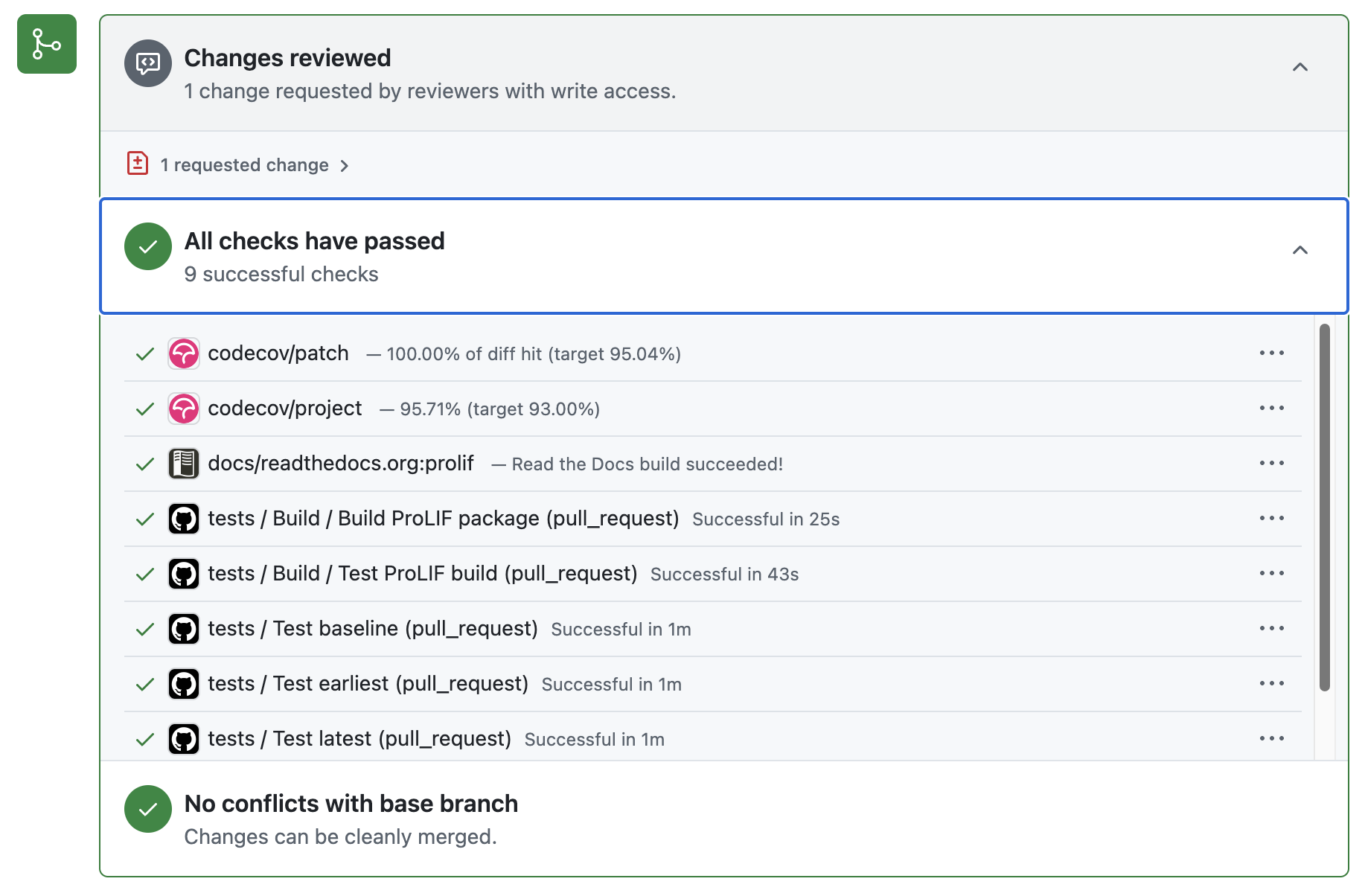
Github bots help you check test coverage and type hints
For the code/linter style checking and type hints, they are routine tasks and can be automated with Github bots. Whenever I push a new commit, the bot runs these checks again and generates reports.
The report of the coverage of test functions.

The report of the coverage of type hints.

Automatic run whenever a new commit is pushed.
Summary
Code review is a crucial part for open source code development. You will find some different opinions from different reviewers. You can explain more about your design and provide a strong rebuttal, or you can accept suggestions for modifying your code. All you need to know is:
Everyone is making the package better.
So, don’t be upset if you have a lot of suggestions during code review. Be patient and sort it out!
See you on the next blog post.
More about GSoC 2025: MDAnalysis x ProLIF project 5
See how we manage this project: click on this [link].
Know more about the project details via GSoC Project Page and ProLIF Github.
Note: The top picture was generated by GPT Image 1 with Japaness Anime style, aspect ratio: 3:2, generation mode: High performance, generation count 4 (I picked one picutre out of four.), and prompts: “I don’t want any text on the picture. Iwould like a drawing with below keywords: Inverting roles between standardization and initialization, protein helper function, coding for cheminformatics, molecule, interaction fingerprint, fixing bond orders of a molecule”.