![Protein helper function fixed your bond orders of residues (molecules) [GSoC 2025 - Week 4]](/images/blog_pics/gsoc-wk-4.JPG)
You may have known that I am a GSoC (Google Summer of Code) contributor in 2025, focusing on “MDAnalysis x ProLIF Project 5: H-Bond interactions from implicit hydrogens” (Intro for this project).
Before developing the main function of calculating implicit H-bond interaction, one important thing need to be prioritized: Make your the structure is correct.. This includes two parts: (1) checking naming convention and standardize the name for residues and (2) fixing bond orders of the molecule.
Residues? Naming convention? Potential issue?
Residue is a substructure of a protein molecule. In general, 20 common amino acids act as residues in a protein.
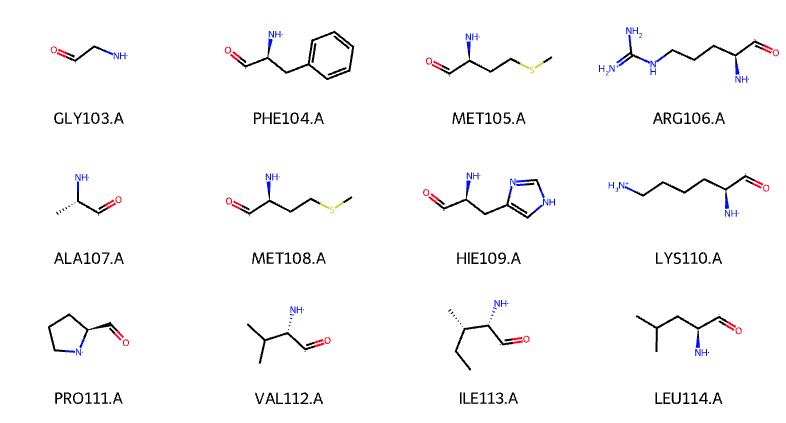
However, some residues have different protonated states. For example, histidine can be protonated on different atoms (proton on ND1 (formal charge: 0), NE2 (formal charge: 0) or both (formal charge: +1)). As a result, various force fields used different names to distinguish the protonated states for the residue. Naming convention for each force field are different (see below table).
| Residue | CHARMM | AMBER | GROMOS | OPLS-AA | Standard name in RDKit |
|---|---|---|---|---|---|
| HIS: proton on ND1 | HSD | HID | HISA | HISD | HIS |
| HIS: proton on NE2 | HSE | HIE | HISB | HISE | HIS |
| HIS: proton on ND1 and NE2 | HSP | HIP | HISH | HISH | HIS |
What bond orders need to be fixed?
Due to the various name of a residue with different protonated states, the structure is not always read as you expected. For instance, RDKit cannot correctly recognize the bond orders of your residues with non-standard name. Here is an example with HSD for histidine (proton on ND1).
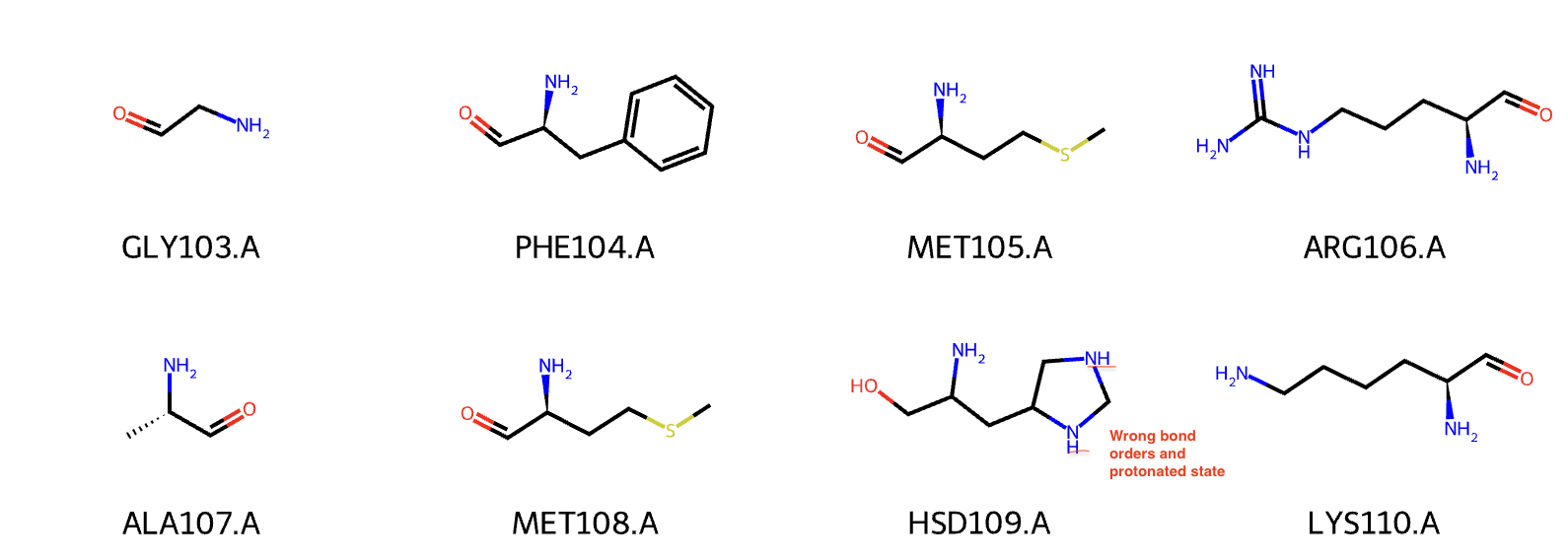
You may know that ProLIF is fundamentally based on RDKit. If the structure is not read correctly, the fingerprint (interaction) will be calculated inaccurately.
Thus, all you need is a protein helper function.
Implementation of protein helper function
The idea of the protein helper function is to use a well-defined template for amino acids (or custom template for special residue) to fix the bond orders. At the same time, the residue names will be standardized.
Here is the pseudo code:
# pseudo code for protein helper function
class ProteinHelper:
Input topology: PDB file path or prolif Molecule
Input custom templates: templates
Main function:
1. Read the molecule as prolif Molecule
2. Guess the forcefield (deal with naming convention) -> guessed forcefield
3. for each residue in prolif Molecule:
[1] get the standardized residue name from a conventional residue name dictionary (with guessed forcefield)
[2] set the residue name with the standardized one
[3] check residue name with templates
[4] fix bond orders with templates
4. Update the protein molecule with bond-order-corrected residues
Output: prolif Molecule with fixed bond orders
Here, we accept two types of templates for fixing bond orders: CIF template and SMILES template. In default, amino acids’ CIF templates are used. However, if the user provides SMILES templates for a residue name, the SMILES templates are prior to the CIF templates when fixing bond orders.
CIF template
The idea using CIF template is come from PDBInf. PDBInf allows you to read PDB files using RDKit and assign bond orders from .cif (PDBx) template files. It is noted that PDBx/mmCIF format is a next-generation file format for storing the informaiton of protein/biomolecule structure. A CIF template of a ligand can be found on RCSB PDB website. For example: Benzamidine.

The file defines atom name for each atom and bond type (single, double, triple, aromatic etc.) for each bond. Thus, the original bond orders in the topology file can be removed and replaced with the correct bond orders.
SMILES template
An alternative and simplier way is to use SMILES as a template. In RDKit, a function rdkit.Chem.AllChem.AssignBondOrdersFromTemplate(refmol, mol) can be used to fix the bond orders (ref). Combining this function with rdkit.Chem.MolFromSmiles(), we can fix bond orders with SMILES templates.
# An example from rdkit:
# rdkit.Chem.AllChem.AssignBondOrdersFromTemplate(refmol, mol)
import os
from rdkit.Chem import AllChem
# read template
template = AllChem.MolFromSmiles("CN1C(=NC(C1=O)(c2ccccc2)c3ccccc3)N")
mol = AllChem.MolFromPDBFile(os.path.join(RDConfig.RDCodeDir, 'Chem', 'test_data', '4DJU_lig.pdb'))
len([1 for b in template.GetBonds() if b.GetBondTypeAsDouble() == 1.0])
# 8 (number of double bonds in the template)
len([1 for b in mol.GetBonds() if b.GetBondTypeAsDouble() == 1.0])
# 22 (number of double bonds in the topology)
# assign correct bond orders
newMol = AllChem.AssignBondOrdersFromTemplate(template, mol)
len([1 for b in newMol.GetBonds() if b.GetBondTypeAsDouble() == 1.0])
# 8 (number of double bonds in the corrected topology)
Example of usage of a protein helper function
Let’s see an example how residues are corrected with protein helper function.
from prolif.datafiles import datapath
from prolif.io.protein_helper import ProteinHelper
# read the protein pdb
protein_path = datapath / "implicitHbond/1s2g__1__1.A_2.C__1.D/receptor_hsd.pdb"
protein_helper = ProteinHelper(input_topology=protein_path)
# standardize the protein (standard amino acid CIF template is used in default)
protein_helper.standardize_protein()
# display the residues
plf.display_residues(protein_helper.protein_mol, slice(100, 108))
# "protein_helper.protein_mol" is the standardized molecule
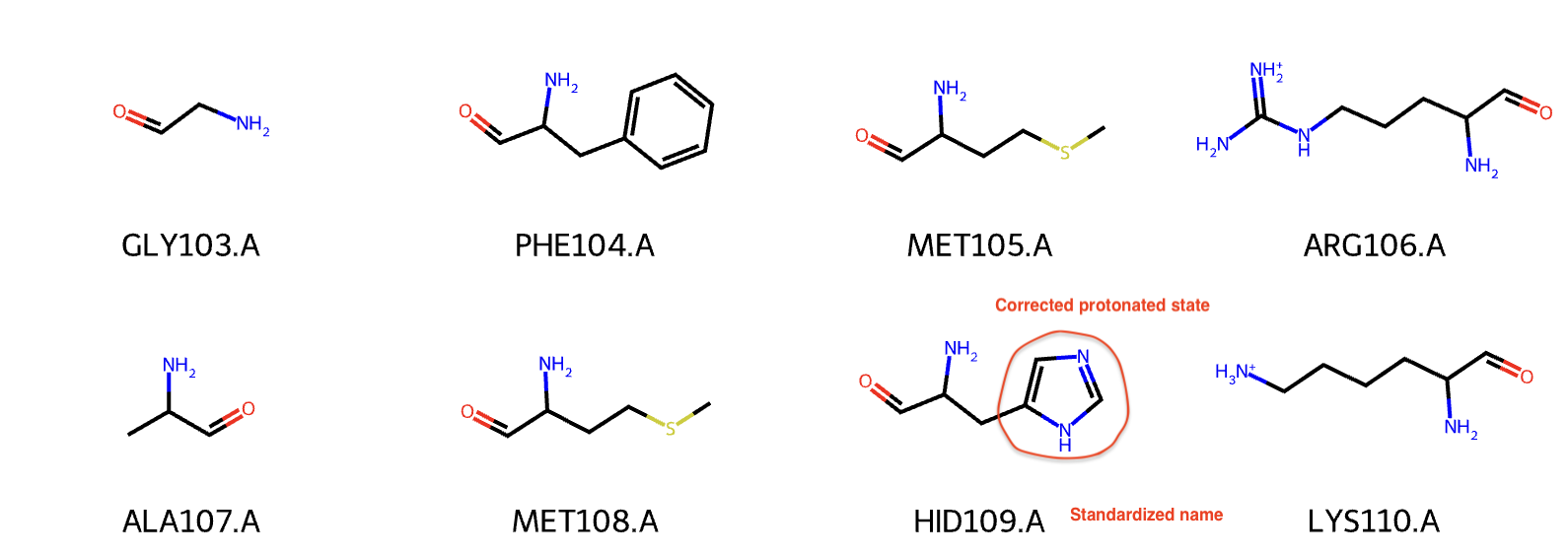
Previously, the histidine (HSD109.A) had wrong bond orders and protonated states. After the standardization in protein helper function, this histidine is now renamed to HID109.A and the bond orders are fixed (proton at the ND1).
Summary
In this blog post, we show the importance of protein helper function. This function standardizes residue names and assigns correct bond orders for residues, preventing potential inaccuracies in the calculation of implicit H-bond interactions.
Next, the main function for calculating implicit H-bond interactions is ready to be developed.
See you on the next blog post.
More about GSoC 2025: MDAnalysis x ProLIF project 5
See how we manage this project: click on this [link].
Know more about the project details via GSoC Project Page and ProLIF Github.
Note: The top picture was generated by Stable Diffusion 3.5 with Pixel Art style, aspect ratio: “16:9” and prompts: “protein helper function, coding for cheminformatics, molecule, interaction fingerprint”.